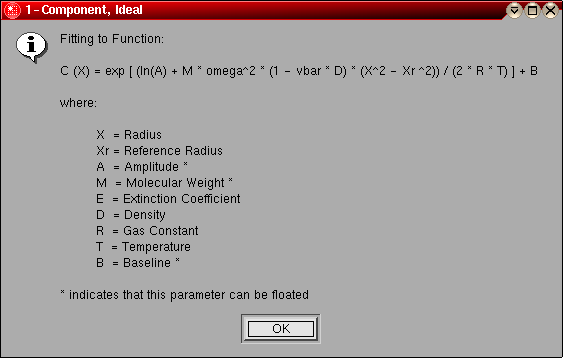
- 1-Component, Ideal: This model can be used for
a pure, single component system that behaves hydrodynamically ideal. This model
should be used first, if model fits well, the system is homogeneous and
doesn't self-associate. The following parameters are fitted:
- A single molecular weight (global)
- The log of the amplitude of the molecular weight term for each
included scan (local)
- The baseline for each included scan (local)
The model number for this model is "0".
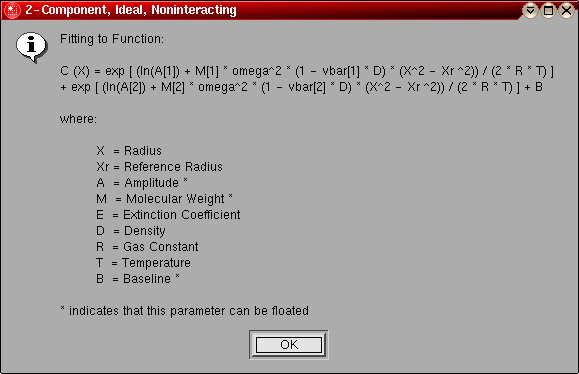
- 2-Component, Ideal, Noninteracting:
This model can be used for for a system of two components that
are not interacting, and behaving hydrodynamically ideal. After
fitting this model, and a good fit was obtained, it is a good idea to
compare the ratios of the integrals between scans run under different
conditions.
If the ratios are roughly unchanged from scan to scan (by an order of
magnitude), there is a good chance that the system is truly noninteracting
and two different components are in the system. If the ratios change
in the direction of larger amounts for the larger component with
increasing concentration and speed, the model should be fitted with a
self-associating model, since the molecular weight average changes with
concentration distribution, and a concentration-dependent self-association
is most likely present. The following parameters are fitted:
- The molecular weight for component 1 (global)
- The molecular weight for component 2 (global)
- The log of the amplitude of molecular weight term 1 for each included
scan (local)
- The log of the amplitude of molecular weight term 2 for each included
scan (local)
- The baseline for each included scan (local)
The model number for this model is "1".
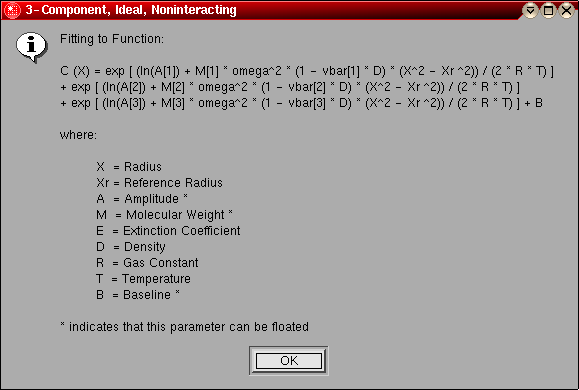
- 3-Component, Ideal, Noninteracting:
This model can be used for for a system of three components that
are not interacting, and behaving hydrodynamically ideal. After
fitting this model, and a good fit was obtained, it is a good idea to
compare the ratios of the integrals between scans run under different
conditions. If the ratios are roughly unchanged from scan to scan
(by an order of magnitude), there is a good chance that the system is
truly noninteracting and three different components are in the system.
If the ratios change in the direction of larger amounts for the larger
components with increasing concentration and speed, the model should be
fitted with a self-associating model, since the molecular weight average
changes with concentration distribution, and a concentration-dependent
self-association is most likely present.
Because this model has a large number of degrees of freedom, this model
should only be used for cases where all other models fail to describe
the system. Rarely do equilibrium scans contain enough information
to quantitatively describe a three component system with all of its
parameters floated. However, if you do know the molecular weight of
each species, you can leave those parameters fixed and just fit the
amplitudes and baseline. However, this would be a linear fit, and is more
appropriately dealt with in the next model, the fixed molecular weight
distribution, described below. The following parameters are fitted:
- The molecular weight for component 1 (global)
- The molecular weight for component 2 (global)
- The molecular weight for component 3 (global)
- The log of the amplitude of molecular weight term 1 for each included
scan (local)
- The log of the amplitude of molecular weight term 2 for each included
scan (local)
- The log of the amplitude of molecular weight term 3 for each included
scan (local)
- The baseline for each included scan (local)
The model number for this model is "2".
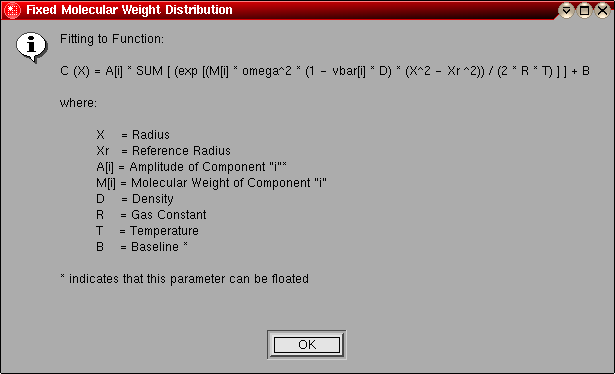
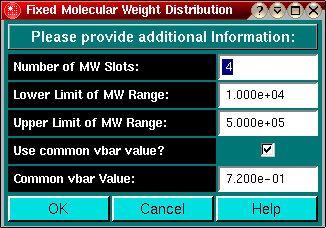
- Fixed Molecular Weight Distribution:
In this model, the molecular weights are preset as an evenly
divided distribution of molecular weights between some lower and upper
molecular weight limit.
This model
can be used for any experiment, since it fits the experiment in an almost
model-independent way. Several diagnostic plots are provided with this model
to ascertain what nonlinear model may be most appropriate for fitting.
Fitting with this model can prove helpful for cases where you need to
distinguish between self-associating and noninteracting systems.
A predetermined molecular weight distribution with the molecular weight
parameter kept fixed is used to fit all scans independently with general
least squares. Since no exponents are fitted, the fitting function can
be considered a linear combination of nonlinear terms, which is linear
in the coefficients that are fitted. The coefficients are nothing more
than the amplitudes for each exponential term with a different fixed
molecular weight. Use at least 3 different molecular weight terms to
describe a model-independent system.
If the residuals for this fit do not come out perfectly random and show
systematic drift, the molecular weight distribution is not set up correctly.
Perform a single component, ideal fit first, and repeat the fixed molecular
weight distribution model by centering your molecular weight distribution
around the molecular weight obtained by the single component fit.
After the fit completes with satisfactory residuals, you can display the
results in one of three ways:
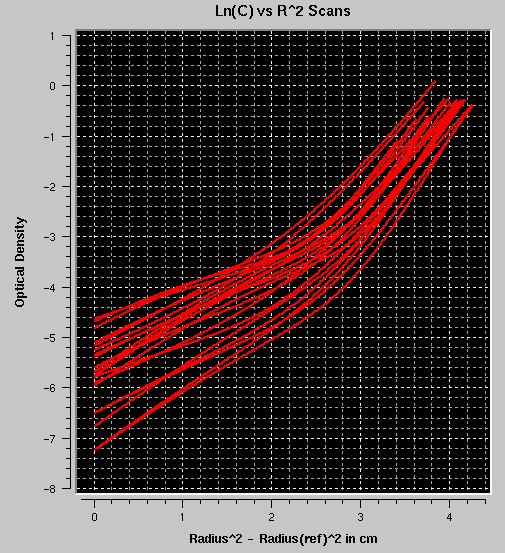
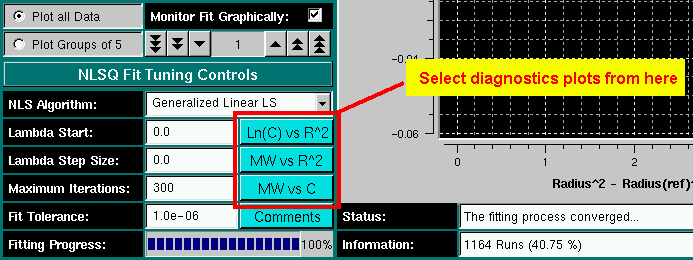
- ln(C) vs. r2: Plots the log
of the concentration of each fit versus the square of the radius. If
multiple components are present in appreciable amounts, the plots will
not be linear. The example shown here reflects a self-associating
monomer-dimer system.
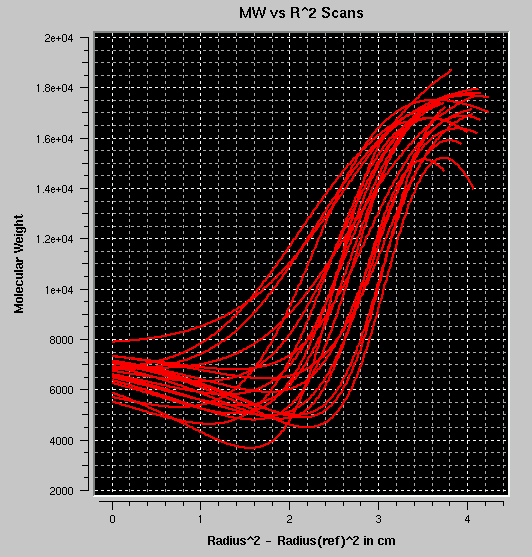
- MW vs. r2: Plots the molecular
weight for each fit at each point versus the square of the radius. If the
plots roughly overlay, the sample is most likely non-interacting. If the
plots follow roughly the sample profile, but each plot at a different
position, the sample is most likely self-associating. If the plots
have multiple inflection points, multiple species are probably present.
The example shown reflects the same self-associating monomer-dimer model
as shown above.
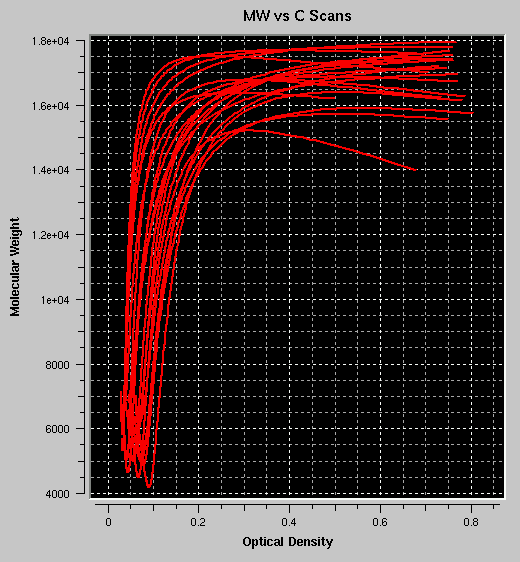
- MW vs. C: Plots the molecular weight for each
fit at each point versus the concentration at that point. If the plots
roughly overlay, the sample is most likely following a concentration
dependent self-association process. If the plots follow roughly the
sample profile, but each plot at a different position, the sample is most
likely non-interacting and heterogeneous in composition. If the plots
have multiple inflection points, multiple species are probably present.
The example shown reflects the same self-associating monomer-dimer model
as shown above.
The plotting functions listed above are available for this model only
and can be accessed from the fitting control panel.
The following parameters are fitted:
- The amplitudes for each molecular weight term for each included scan (local)
- The baseline (or zeroth order term) for each included scan (local)
The following parameters are fixed:
- Each molecular weight term (global)
The model number for this model is "3".
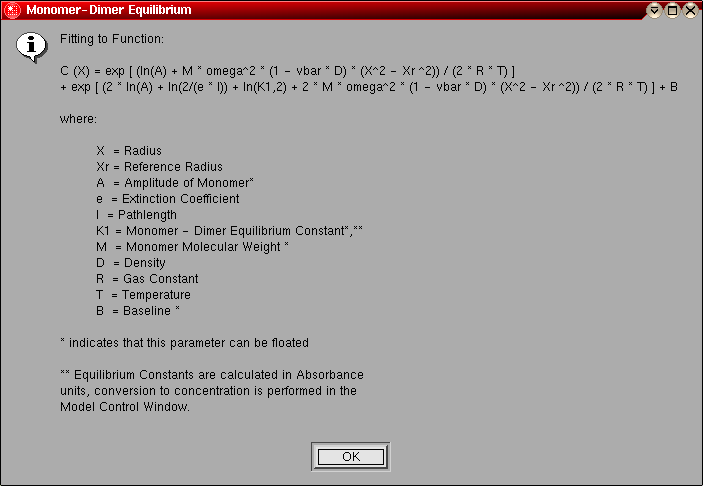
- Monomer-Dimer Equilibrium: This model can be
used to fit an ideal, reversibly self-associating monomer-dimer system.
The following parameters are fitted:
- The monomer molecular weight (global)
- The log of the amplitude of the monomer molecular weight term for
each included scan (local)
- The log of the monomer-dimer association constant (global)
- The baseline for each included scan (local)
The model number for this model is "4".
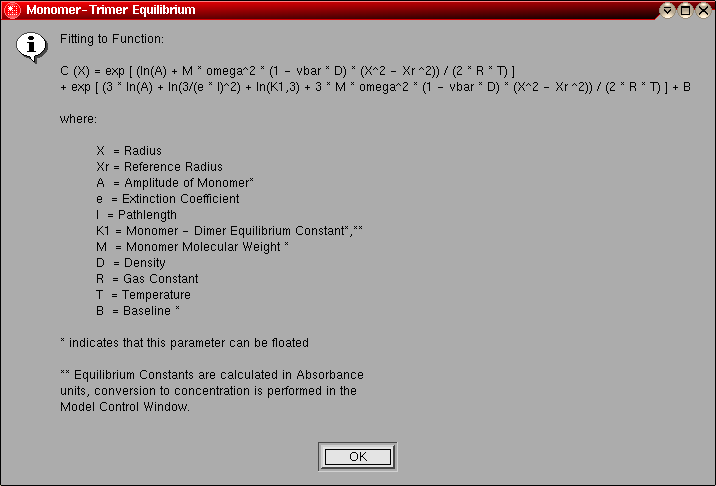
- Monomer-Trimer Equilibrium: This model is
identical to the monomer-dimer equilibrium model, except instead of a dimer
the association is for a trimer.
The model number for this model is "5".
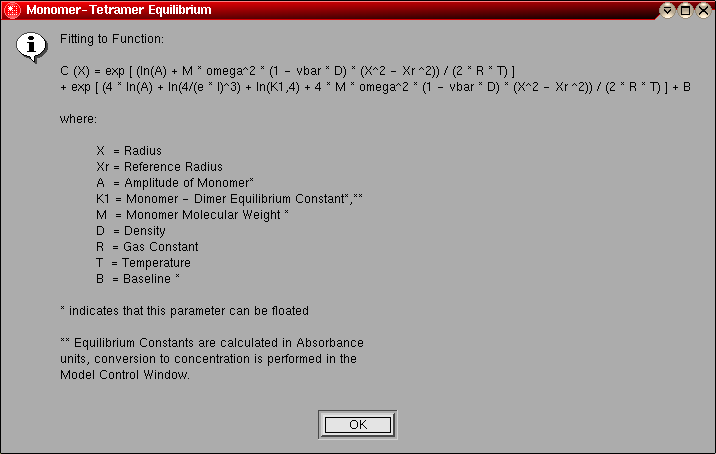
- Monomer-Tetramer Equilibrium: This model is
identical to the monomer-dimer equilibrium model, except instead of a dimer
the association is for a tetramer.
The model number for this model is "6".
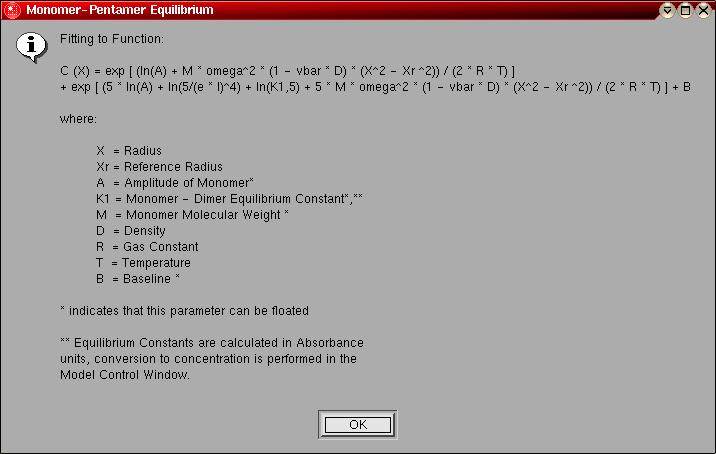
- Monomer-Pentamer Equilibrium: This model is
identical to the monomer-dimer equilibrium model, except instead of a dimer
the association is for a pentamer.
The model number for this model is "7".
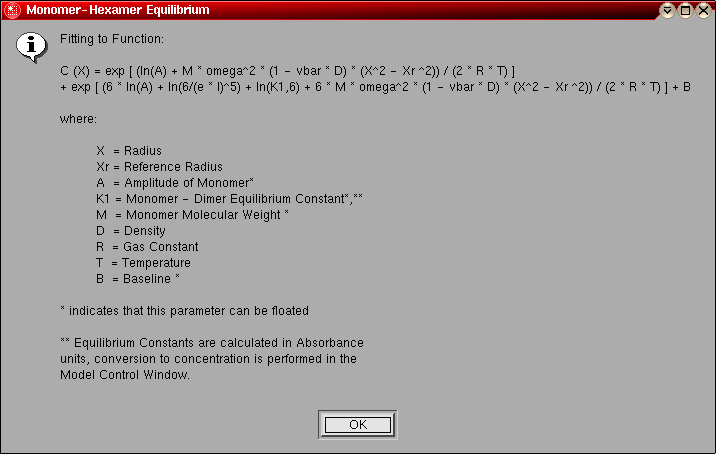
- Monomer-Hexamer Equilibrium: This model is
identical to the monomer-dimer equilibrium model, except instead of a dimer
the association is for a hexamer.
The model number for this model is "8".
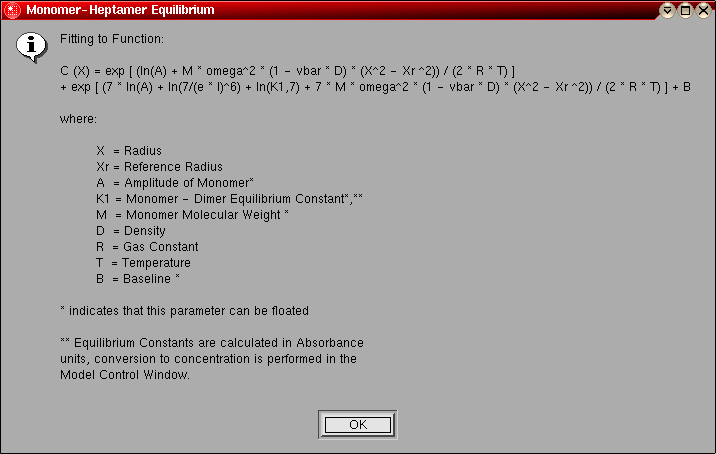
- Monomer-Heptamer Equilibrium: This model is
identical to the monomer-dimer equilibrium model, except instead of a dimer
the association is for a heptamer.
The model number for this model is "9".
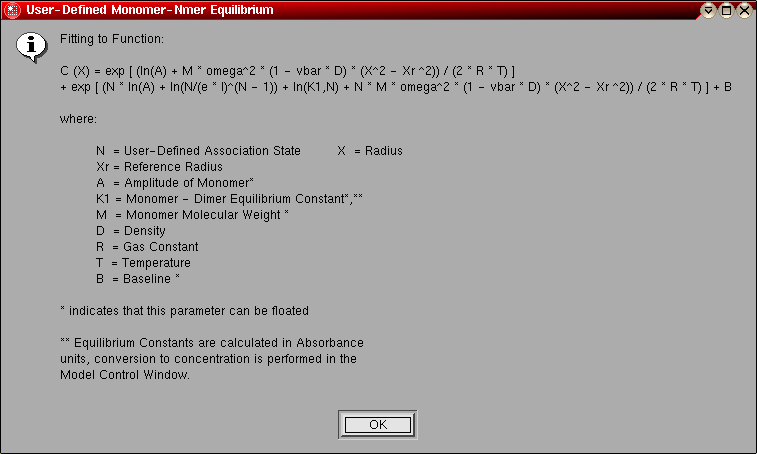
- User-defined Monomer-Nmer Equilibrium:
This model is identical to the monomer-dimer equilibrium model,
except instead of a dimer the association is for a user-defined
stoichiometry. You can define your desired stoichiometry in the
monomer-Nmer stoichiometry selection panel.
The model number for this model is "10".
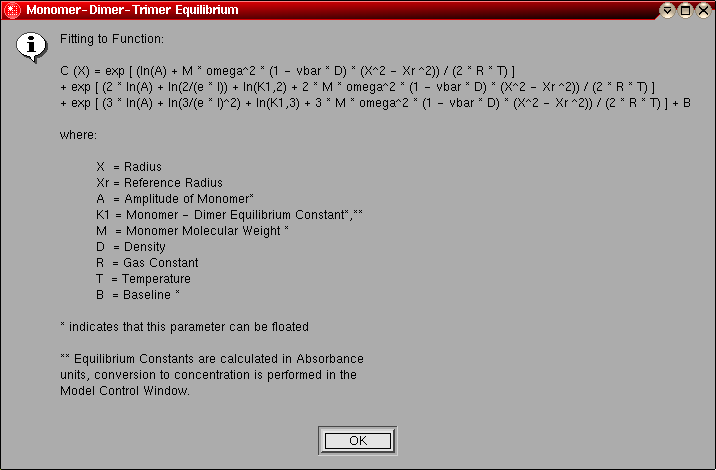
- Monomer-Dimer-Trimer Equilibrium:
This model can be used to fit an ideal, reversibly self-associating
monomer-dimer-trimer system. The following parameters are fitted:
- The monomer molecular weight (global)
- The log of the amplitude of the monomer molecular weight term for
each included scan (local)
- The log of the monomer-dimer association constant (global)
- The log of the monomer-trimer association constant (global)
- The baseline for each included scan (local)
The model number for this model is "11".
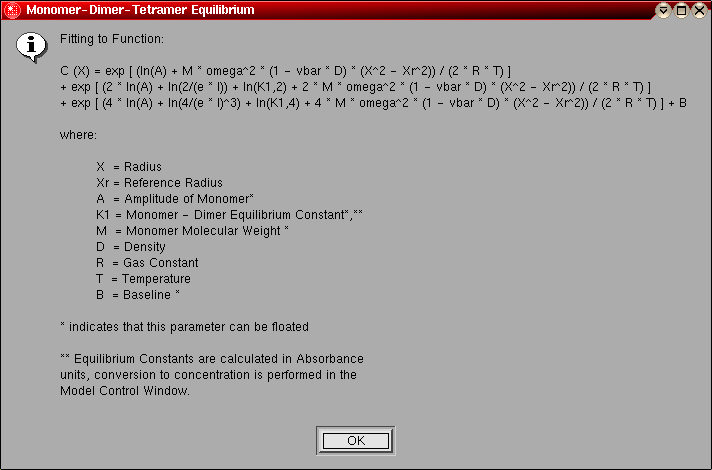
- Monomer-Dimer-Tetramer Equilibrium:
This model can be used to fit an ideal, reversibly self-associating
monomer-dimer-tetramer system. The following parameters are fitted:
- The monomer molecular weight (global)
- The log of the amplitude of the monomer molecular weight term for
each included scan (local)
- The log of the monomer-dimer association constant (global)
- The log of the monomer-tetramer association constant (global)
- The baseline for each included scan (local)
The model number for this model is "12".
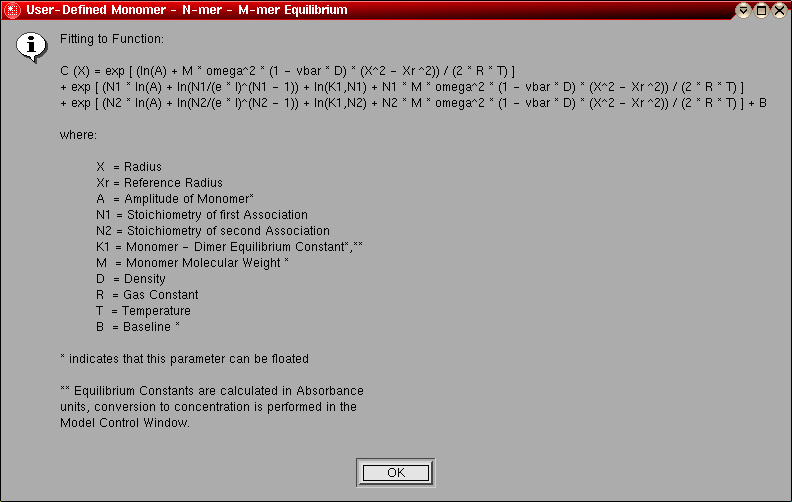
- User-defined Monomer - N-mer - M-mer
Equilibrium: This model can be used to fit an ideal, reversibly
self-associating monomer - N-mer - M-mer system. You can define the
stoichiometries for the N and M associations using the
stoichiometry selection panel. The following
parameters are fitted:
- The monomer molecular weight (global)
- The log of the amplitude of the monomer molecular weight term for
each included scan (local)
- The log of the monomer - N-mer association constant (global)
- The log of the monomer - M-mer association constant (global)
- The baseline for each included scan (local)
The model number for this model is "13".

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}